Rosetta, the macromolecular software modeling suite (which I contribute to as a developer), is great at modeling proteins, but not always so great at dealing with non-protein chemistry, including small molecules.
I've worked on several projects that used Rosetta with the intent of designing a protein to bind a novel small molecule. At the beginning of the workflow, I usually download a two-dimensional representation of the small molecule that, when visualized, looks something like this:
Rosetta needs to be given a parameter file specifying the chemistry of a small molecule in order to score it. Recent design algorithms such as coupled moves [@@ollikainen_coupling_2015] also require input of "conformers", a set of low-energy, three-dimensional states that the molecule is likely to access. Appropriate conformer input, when visualized, looks like this:
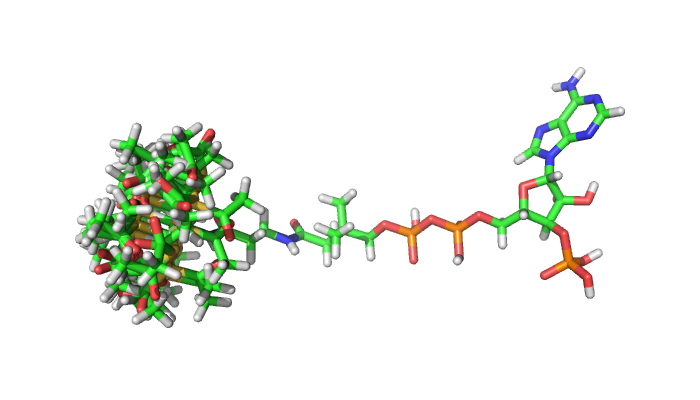
Note that since acetyl-CoA is a large (small) molecule, the conformers I generated only access rotamer degrees of freedom at one end of the acetyl-CoA molecule. If I did not limit degrees of freedom in this way, the number of conformers to generate and then sample in Rosetta would be intractable. Additionally, if I were going to design a catalytic interaction between acetyl-CoA and a protein of interest, only the portion of the small molecule that is near the active site would need to be modeled as flexible in a coupled moves design simulation.
As my lab does not currently license any of the commercially available software (which can be quite expensive) commonly used to generate small molecule conformers, I needed to find free for academic use(and preferably open source) software to do the job. Fortunately, several good options exist, such as the Confab [@@oboyle_confab_2011] algorithm within the Open Babel [@@oboyle_open_2011] chemical toolbox. Even better, the performance of Confab has been compared with other open-source and paid software [@@ebejer_freely_2012], and it compares favorably, especially when used on flexible molecules (those with 10 or more rotatable bonds).
I then ran into a snag: I couldn't find an off-the-shelf way with Confab's command line interface to generate conformers/rotamers for only a subset of rotatable bonds in a small molecule. Fortunately, Open Babel has powerful bindings for scripting available, including in Python. I wrote a script that takes in a small molecule as input and an optional argument of atoms allowed to move in conformer generation. Rotatable bonds will only be sampled by Confab if they involve these specified "unfrozen" atoms.
You can view the script embedded below, at the gist page at Github with formatting and the ability to comment, or as a raw file ready to download. I hope it's useful! Improvements are welcome as well; I've licensed the script under the MIT license.